At COMPARE.EDU.VN, we understand that interpreting gel electrophoresis results can be challenging. This comprehensive guide offers a step-by-step approach on How To Compare Gel Electrophoresis Results, ensuring accurate analysis and troubleshooting of PCR experiments. This article equips you with the necessary knowledge to interpret electrophoresis gels with confidence, enhance your PCR workflows, and make informed decisions based on your experimental findings. Let’s explore DNA fragment separation, size estimation, and agarose gel electrophoresis interpretation, ensuring your research is backed by solid, comparable data.
1. Capturing and Documenting Electrophoresis Gel Images
The first critical step in interpreting gel electrophoresis results is capturing a clear, high-quality digital image of the gel. This image serves as a permanent record of your experiment and allows for detailed analysis, annotation, and easy sharing.
1.1. Image Acquisition Techniques
- Smartphone or Compact Camera: A smartphone camera or small compact camera can be used to photograph the gel. Adjust the focus manually to ensure the amplicon bands are sharp.
- Gel Imaging Hood: Use a gel imaging hood or improvised dark box to exclude ambient light.
- Bento Lab Gel Tank Lid: Photograph the gel through the Bento Lab gel tank lid in dark conditions.
1.2. Optimizing Image Quality
- Condensation: Ensure the orange filter or orange tank lid is free of condensation. If not, wipe it clean with a lint-free tissue.
- Ambient Light: Reduce ambient light as much as possible. The darker the room, the better the visualization.
- Alternative Method: If excluding ambient light is difficult, carefully remove the gel and place it on a piece of transparent plastic directly on the transilluminator. Cover it with a gel imaging hood or dark box. Remember to wear gloves and change them after handling the gel to avoid potential PCR contamination.
- Gel Map: Photograph a gel map showing the sample arrangement in the wells before you photograph your gel.
1.3. Annotation and Documentation
Properly documenting your electrophoresis results is essential for maintaining a thorough and organized laboratory record. Documenting the images will make it easier to compare results across multiple experiments.
- Transfer Images: Transfer the gel images into an electronic document, such as Google Docs or Slides, or print the images to glue into a physical lab book.
- Annotation: Annotate the gel image with sample codes, results, and your interpretation as part of the recorded experimental process.
- Image Adjustment: Before documenting the gel, adjust the image to enhance clarity. Use phone or desktop apps to crop, rotate, adjust contrast, color saturation, and gamma intensity. You can also invert the image to a white background with dark/black bands.
2. Assessing the Quality of the Agarose Gel
The physical quality of the agarose gel significantly impacts the quality of the gel image and the accuracy of the results. Identifying and addressing issues early can save time and prevent inaccurate interpretations.
2.1. Common Gel Quality Issues
| Gel Quality Issues | Solution |
|---|---|
| Bubbles present in the gel | Allow the gel to cool before casting, enabling bubbles to rise and pop. If bubbles remain, manually remove them using a pipette tip. Remove and replace the comb to prevent bubbles from clinging to the sides. |
| Cloudy agarose | Ensure the agarose is completely dissolved during melting. Bring it to a bubbling state once, swirl, and then bring it to a bubbling state again. |
| Physical objects in the gel (dust, hairs) | Cover the gel tank with a paper towel while open and setting. |
| Gel melted while running | Reduce the running temperature by using a lower voltage and shorter durations. For long, slow runs with low-percentage agarose gels, use a running buffer that allows cooler, faster electrophoresis than TBE buffer (e.g., sodium borate, lithium borate, or lithium acetate borate). |
| DNA stain artifacts | Ensure the DNA stain is well-mixed with the molten agarose. Let it sit to remove bubbles before pouring. |

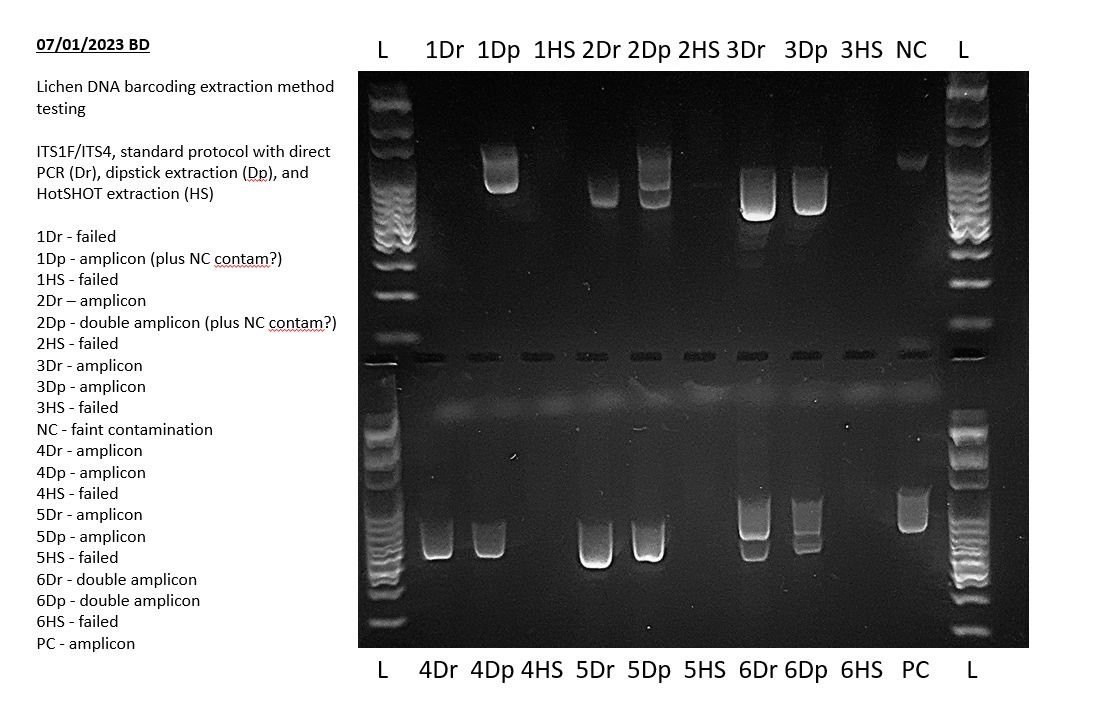
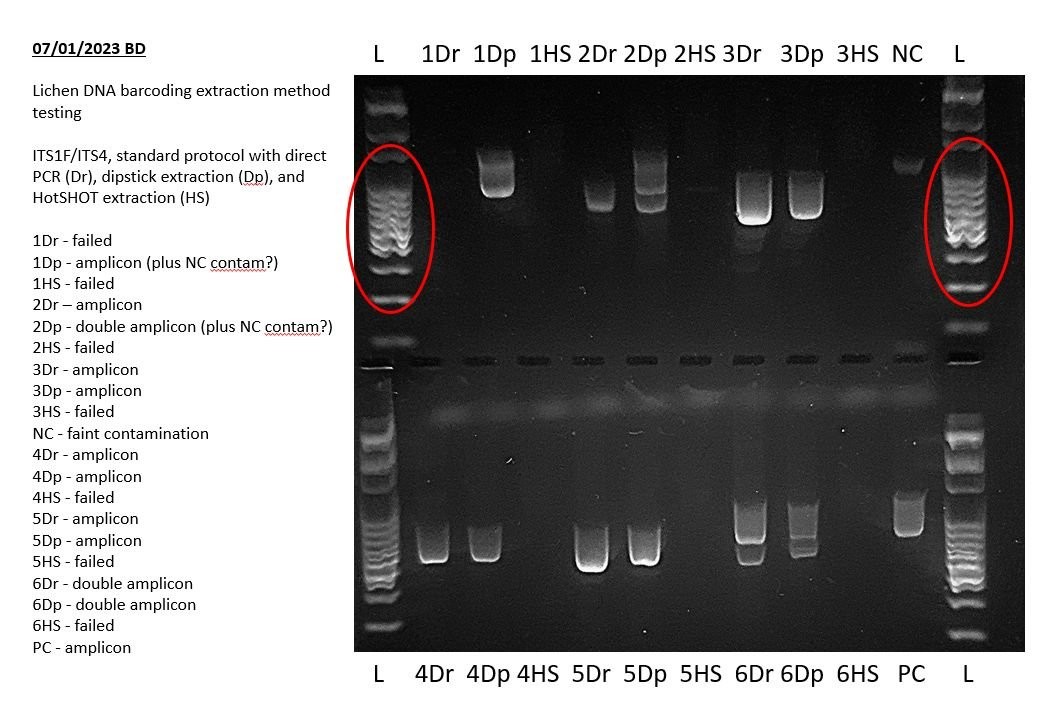
Addressing these issues ensures a solid foundation for accurate gel electrophoresis results and reliable PCR analysis. This attention to detail is vital for anyone looking to compare gel electrophoresis results effectively and interpret their findings with confidence.
3. Evaluating DNA Ladder Performance
The DNA ladder serves as a crucial reference point for estimating the size of DNA fragments in your samples. Evaluating its performance helps determine the overall quality of the electrophoresis run.
3.1. Key Indicators of Ladder Performance
- Visibility: The DNA ladder should have clearly visible bands in a ladder pattern in the first and last lanes, running at least through the top half of the gel. This indicates that the gel has run successfully.
- Clarity and Distinctness: DNA ladder bands should be clear, distinct, and crisp, especially in the size ranges of the target amplicons.
- Uniformity: Ensure the ladder runs straight and evenly across the gel.
3.2. Common Issues and Solutions
| DNA Ladder Running Issues | Problem and Solution |
|---|---|
| The ladder has not run far enough | Run the gel for longer periods. |
| The ladder has smeared | Reduce the amount of DNA loaded. Smearing can also occur with some DNA stains, so consider switching to a different stain. |
| Poor band separation | Run the gel for a longer duration. Optimize the agarose gel percentage for your target amplicon sizes. |
| The ladder is not clearly visible | Ensure correct pipetting and that the ladder settled into the well before running. Verify that DNA stain was added and that the gel has not been overexposed to light. For some DNA stains (like GelGreen), dye precipitation can be reversed by heating the tube in hot water at 45-50°C for several minutes and shaking well. |
| The ladder has run crooked | Ensure the gel was set and run on a level surface using a spirit level. Check and straighten the electrode wires in your gel tank, avoiding damage. Ensure the electrodes are not covered in agarose gel due to leakage past the rubber dams during casting. |
| Ladder in unintended lanes | Pipette carefully to avoid misplacing ladder DNA into other wells or onto the surface of the agarose gel. |
3.3. Understanding the DNA Ladder Composition
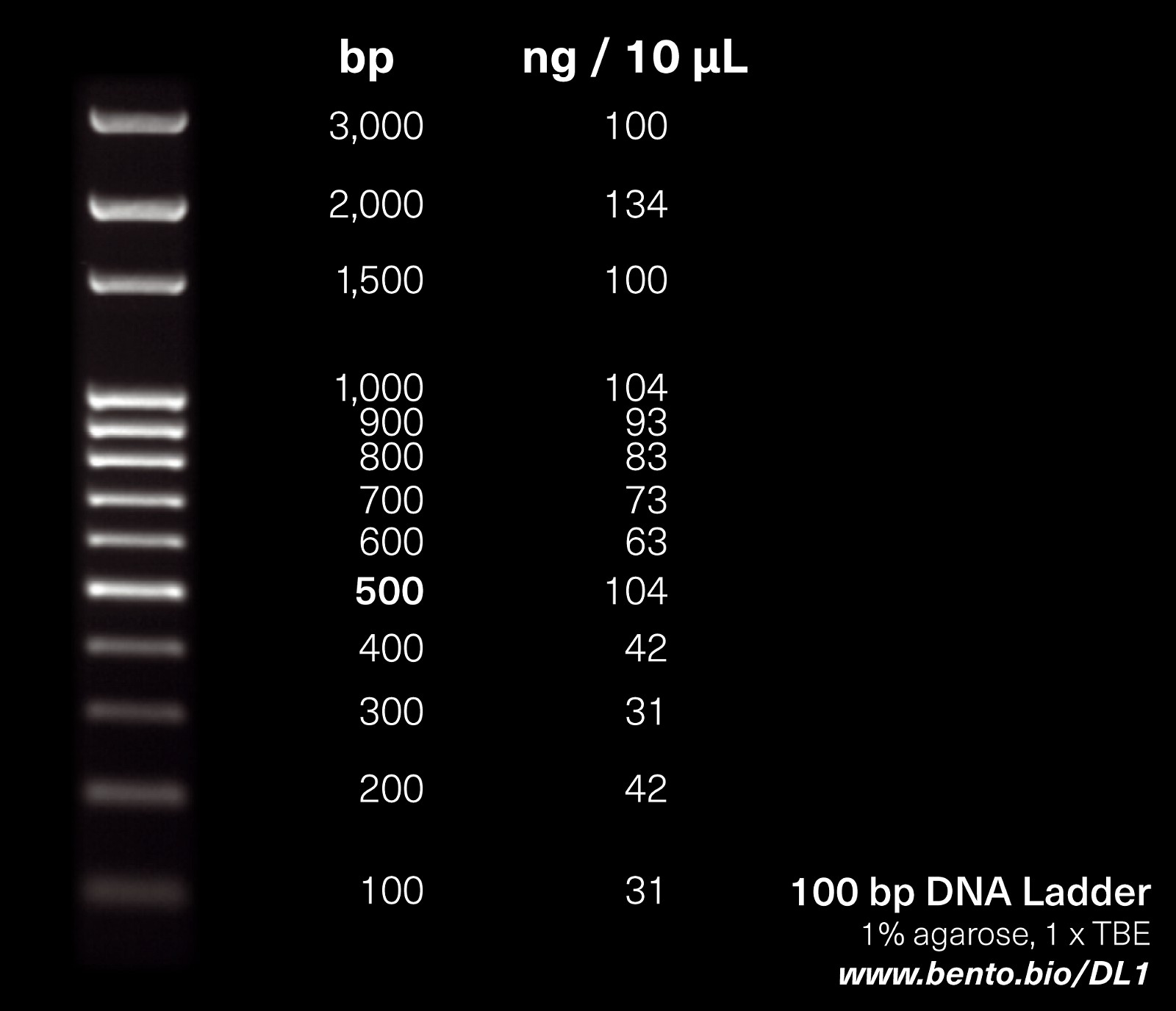
The DNA ladder is a mixture of DNA fragments of known sizes, typically ranging from 100 to 3,000 base pairs (bp). The 100 bp DNA ladder contains DNA fragment sizes ranging from 100 to 1,000 in 100 bp increments, then (in some ladders) 1500, 2000, and 3000. Each band is provided in a particular concentration (ng DNA/unit volume). Consult the product information for your DNA ladder for specifics on the band size and concentrations. The smallest bands are at the bottom of the gel and the larger bands are at the top.
To estimate the size of your PCR amplicon, compare its position to the ladder. For example, if your amplicon is halfway between the 500 bp and 600 bp bands, its size is approximately 550 bp. Running the DNA ladder in the first and last wells, or adjacent to PCR products, improves comparability. A middle lane of ladder can also detect any differences in migration rate.
4. Examining Negative Controls
The negative control is a critical component of PCR experiments used to detect contamination in your PCR reagents or workflow.
4.1. Expected Outcome
Ideally, the negative control lane should be completely blank, indicating no amplification occurred in the absence of template DNA.
4.2. Interpreting Results
- Absence of Bands: A blank negative control confirms that the PCR mix and workflow are free from contamination.
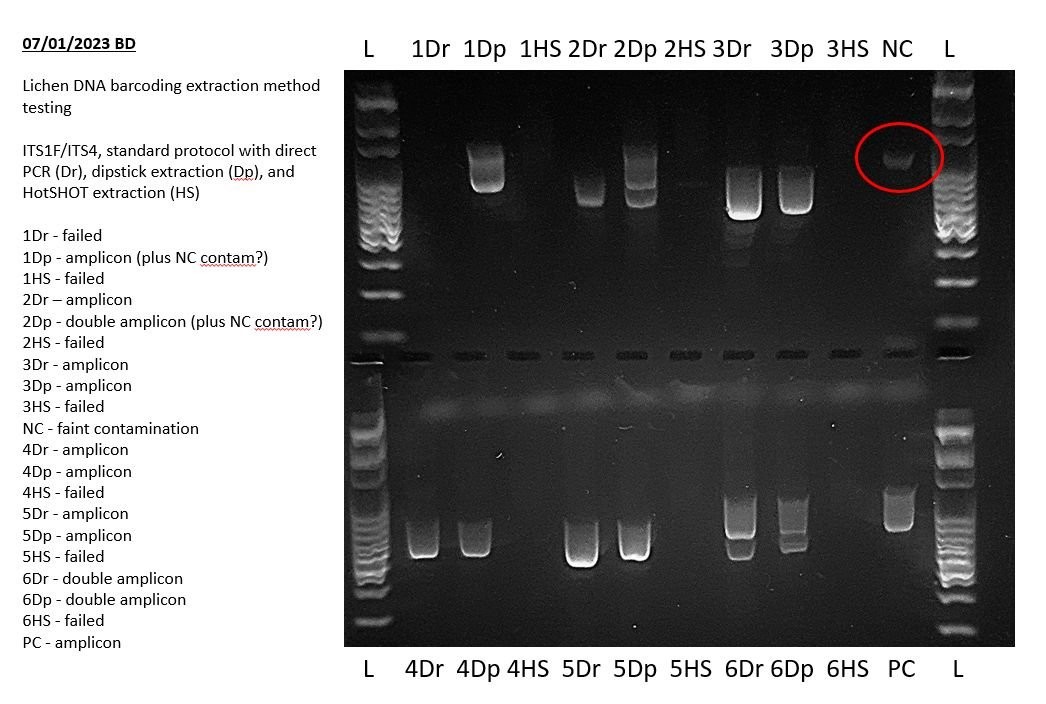
- Presence of Bands: If a band is present in the negative control (other than residual primers at below 100 bp in size), your PCR mix or workflow was contaminated.
The intensity of the band can provide insight into the severity of the contamination. Even a faint band indicates a potential issue that needs to be addressed. If the band is faint and not evident in other PCR products, it may not be a major issue. However, you should still check your reagents and workflow for PCR contamination and decontaminate as appropriate.
4.3. Corrective Actions
If contamination is detected, take the following steps:
- Identify the Source: Carefully examine all reagents and equipment for potential sources of contamination.
- Decontaminate: Thoroughly clean the work area, pipettes, and other equipment with appropriate decontamination solutions.
- Prepare Fresh Reagents: Prepare fresh PCR master mix and other critical reagents.
- Repeat the Experiment: Repeat the PCR with the fresh reagents and a decontaminated workflow to confirm the contamination has been eliminated.
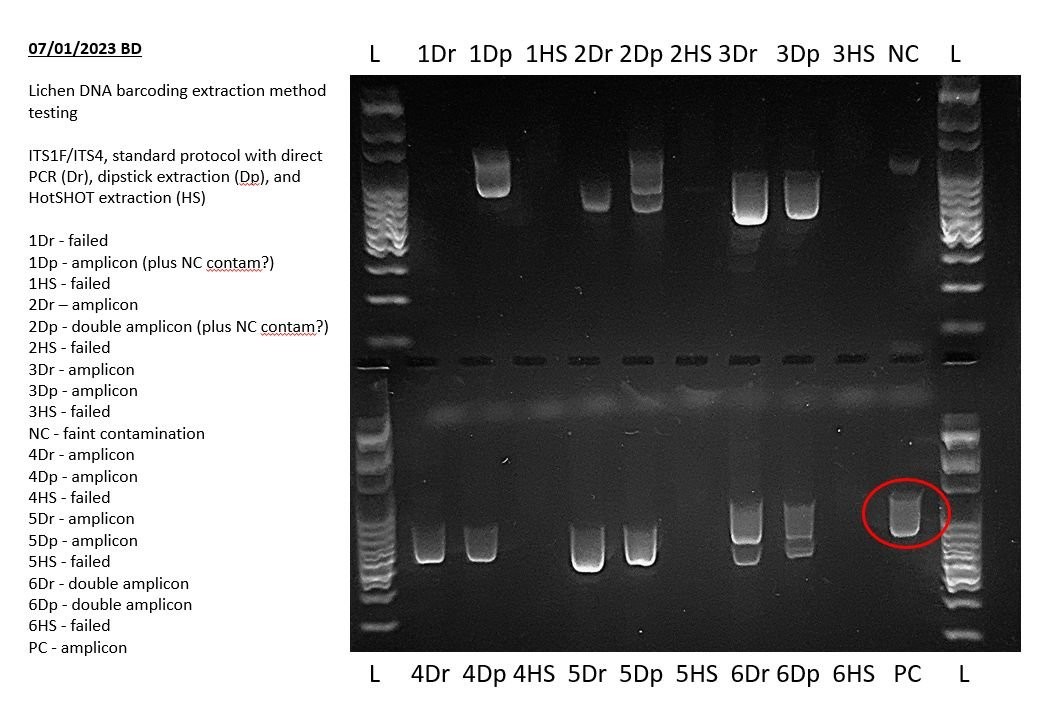
The above example shows a very faint positive result in the negative PCR control during a lichen barcoding exercise. This indicates that some of the results of this experiment may not sequence well and may need to be repeated. Specifically, any bands of the same size in other lanes may indicate that those PCRs also amplified this contaminant sequence.
5. Analyzing Positive Controls
A positive control is a PCR reaction containing a known DNA template that should produce a specific, predictable result.
5.1. Expected Outcome
The positive control lane should display a clear, bright amplicon band of the expected size.
5.2. Interpreting Results
- Successful Amplification: A clear band of the expected size confirms that the PCR conditions, reagents, and equipment are functioning correctly.
- Failed Amplification: If the positive control fails to amplify, it indicates a problem with the PCR setup or reagents.
5.3. Troubleshooting
If the positive control fails, consider the following troubleshooting steps:
- Verify Reagents: Ensure all reagents are fresh, properly stored, and not expired.
- Check PCR Conditions: Review the PCR protocol to ensure the correct cycling parameters are being used.
- Confirm Primer Design: Verify that the primers are specific to the target sequence and that they are free from secondary structures or primer-dimer formation.
- Replace Reagents: If necessary, replace critical reagents such as the DNA polymerase or master mix.
If the positive control failed, and all your sample PCRs failed, then that suggests an issue with the PCR.
For more information on controls, you can consult our article on Controls in PCR and PCR assays with Bento Lab.
6. Evaluating Sample PCR Results
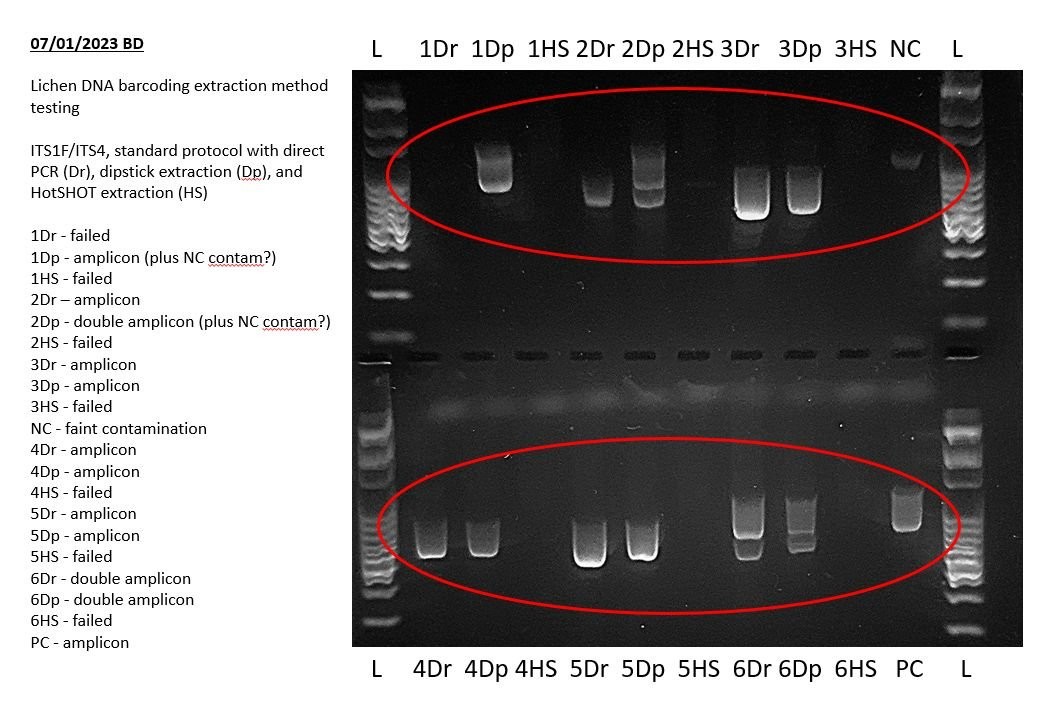
Evaluating sample PCR results involves analyzing the presence, size, and quality of amplicons in each sample lane. This step helps determine whether the PCR was successful and whether the results align with expectations.
6.1. Expected Results
Before starting your experiment, you should have a clear expectation of the PCR results, such as:
- A single amplicon band of a specific size
- Multiple amplicon bands of different sizes
- A range of amplicon band sizes
6.2. Common Artifacts
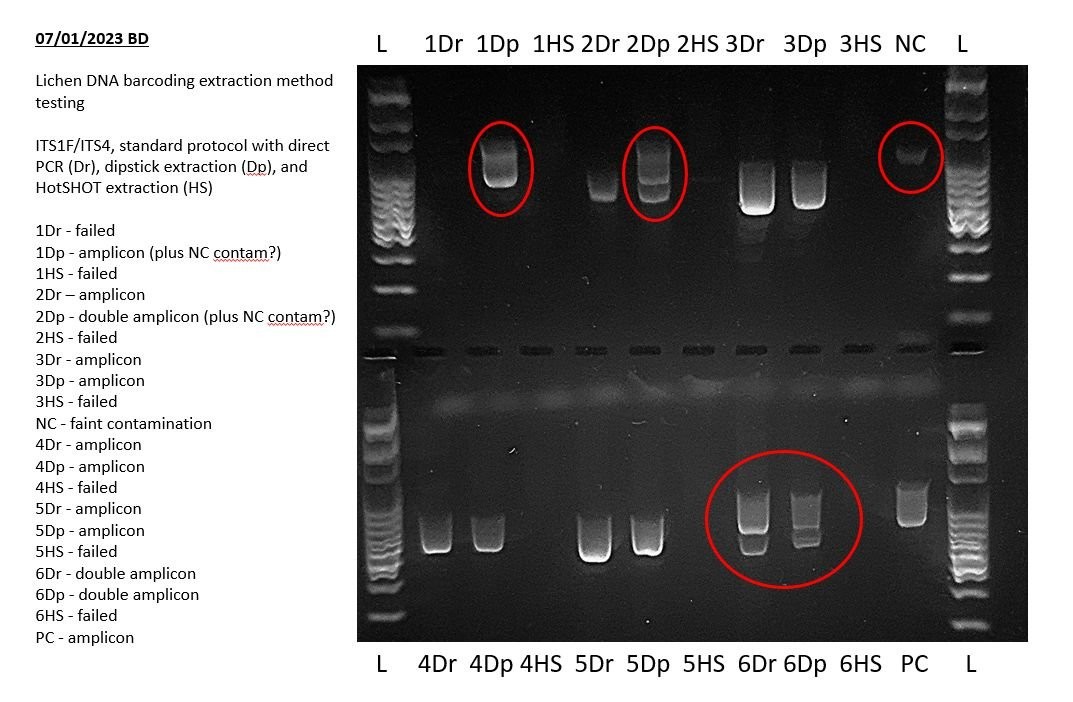
- Primer Dimers: Bright bands of under 100 bp in size are likely residual primers or primer dimers, unless these are expected as part of your PCR assay.
- Non-Specific Amplification: Larger unexpected bands are likely non-specific amplification, especially if they contain long smears instead of discrete bands.
- Stuck PCR Products: PCR products that have entirely or partially stuck in wells, accompanied by smears, may be caused by non-specific amplification or by physical or chemical obstacles to migration.
6.3. Troubleshooting Failed Samples
If most of your samples worked as expected but some didn’t, then the ones that didn’t may not have amplified due to either insufficient template DNA present or PCR inhibition caused by residual PCR inhibitors from the DNA extraction. If most of your samples were of the same subsample size and in good condition, then PCR inhibition may be more likely, and the extract may need dilution before use as a DNA template in PCR.
If you have no successes at all, then you can use your positive control to identify if it was a systemic PCR problem. If the positive control failed then there was probably a PCR issue. If the positive control was successful there was probably a DNA extraction issue with your samples.
7. Assessing Uneven Gel Running
Uneven running of the gel can distort band migration, leading to inaccurate size estimations and misinterpretation of results.
7.1. Identifying Uneven Running
- Crooked Lanes: The PCR products and DNA ladder may run slightly askew rather than straight across the gel.
- Variable Migration: Middle lanes may run faster than those at the edge.
7.2. Causes of Uneven Running
- Mini-Gel Limitations: Small mini-gel tanks and gels are more prone to uneven running.
- Running Speed: Running gels faster than recommended can exacerbate uneven running.
- Misaligned Electrodes: Gel tank electrodes may become misaligned.
7.3. Corrective Measures
- Lower Voltage: Run the gel at a lower voltage for a longer time.
- Electrode Alignment: Check and straighten gel tank electrodes, taking care not to damage them.
- Level Surface: Ensure the gel is set and run on a level surface.
8. Detecting Missing Bands in Multiplex PCR Assays
In multiplex PCR, multiple amplicons are targeted in a single reaction. The absence of expected bands can indicate PCR bias or allelic dropout.
8.1. Understanding PCR Bias
PCR bias occurs when multiple amplicons in a PCR exhibit different amplification efficiencies, resulting in unequal band intensities.
8.2. Identifying Allelic Dropout
Allelic dropout is a phenomenon where one of two gene variants (alleles) fails to amplify, leading to a false result of homozygosity.
8.3. Minimizing Band Loss
- Optimized Assays: Well-designed PCR assays are experimentally optimized to reduce PCR biases before publication.
- Standardized Procedures: Follow PCR protocols precisely to minimize variability.
9. Identifying Unexpected Bands, Primer Dimers, and PCR Artifacts
Unexpected bands, primer dimers, and other artifacts can complicate the interpretation of gel electrophoresis results.
9.1. Potential Causes
- Contamination: Systemic contamination can introduce unwanted DNA templates into the PCR.
- Non-Specific Amplification: Non-specific primer binding can result in the amplification of unintended DNA fragments.
- Primer Dimers: Primer dimers form when primers bind to each other instead of the target DNA sequence.
9.2. Comparison with Controls
Compare any additional bands in your results with your negative control to check for systemic contamination.
9.3. Troubleshooting Non-Specific Amplification
For more information on non-specific amplification, you can read our article on Troubleshooting Non-Specific Amplification with Bento Lab.
10. The Role of COMPARE.EDU.VN in Simplifying Gel Electrophoresis Result Comparison
Understanding and interpreting electrophoresis gels is crucial for assessing the results of your PCRs. A systematic, step-by-step approach allows you to:
- Document and annotate gels
- Assess gel and gel run quality
- Evaluate the success of positive and negative controls
- Analyze results and check for missing or unexpected bands
At COMPARE.EDU.VN, we provide comprehensive resources to simplify the process of comparing gel electrophoresis results. Our detailed guides, expert insights, and user-friendly tools empower you to interpret your experimental data with confidence and make informed decisions.
10.1. Benefits of Using COMPARE.EDU.VN
- Comprehensive Comparisons: Access detailed comparisons of different gel electrophoresis techniques, equipment, and reagents.
- Expert Insights: Benefit from expert analyses and troubleshooting tips to resolve common issues.
- Step-by-Step Guides: Follow our step-by-step guides for accurate and consistent gel electrophoresis analysis.
- Community Support: Connect with other researchers and share your experiences and insights.
Ready to streamline your research? Visit COMPARE.EDU.VN today to discover the resources you need to compare gel electrophoresis results effectively and advance your scientific endeavors.
Address: 333 Comparison Plaza, Choice City, CA 90210, United States
WhatsApp: +1 (626) 555-9090
Website: COMPARE.EDU.VN
Frequently Asked Questions (FAQs) About Gel Electrophoresis Result Interpretation
1. What is gel electrophoresis, and why is it important?
Gel electrophoresis is a technique used to separate DNA fragments based on their size. It is crucial for visualizing amplified DNA, estimating DNA fragment sizes, and assessing DNA concentration.
2. How do I prepare a gel for electrophoresis?
To prepare a gel for electrophoresis, you need agarose powder, a buffer solution (such as TBE or TAE), and a gel casting system. Mix the agarose powder with the buffer solution, heat until the agarose is completely dissolved, and then pour the mixture into the gel casting system with a comb to create wells.
3. What is the purpose of a DNA ladder in gel electrophoresis?
A DNA ladder, also known as a DNA marker, is a mixture of DNA fragments of known sizes. It is used as a reference to estimate the size of DNA fragments in your samples.
4. How do I load my samples into the gel?
Mix your DNA samples with a loading dye, which contains a dense substance (such as glycerol) to help the sample sink into the well, and a tracking dye (such as bromophenol blue) to monitor the progress of the electrophoresis. Carefully pipette the mixture into the wells, avoiding bubbles.
5. What voltage should I use for gel electrophoresis?
The optimal voltage depends on the size and type of gel, as well as the buffer used. A typical voltage range is 80-150V for a mini-gel. Running at a lower voltage for a longer time can improve band resolution.
6. How long should I run the gel?
The duration of the electrophoresis run depends on the size of the DNA fragments you are trying to separate. Monitor the progress of the run by observing the tracking dye. Stop the run when the dye has migrated sufficiently far enough to separate the bands of interest.
7. What are primer dimers, and how can I avoid them?
Primer dimers are small, non-specific products that form when primers bind to each other instead of the target DNA sequence. They appear as small, bright bands at the bottom of the gel. To avoid primer dimers, optimize your primer design, use a hot-start polymerase, and increase the annealing temperature.
8. How do I interpret the results of my gel electrophoresis?
Compare the position of your DNA bands to the DNA ladder to estimate their size. Check the negative control lane for any signs of contamination. Ensure the positive control shows a clear band of the expected size. Analyze the presence, size, and intensity of bands in your sample lanes to determine whether the PCR was successful.
9. What should I do if my gel electrophoresis results are not clear?
If your gel electrophoresis results are not clear, consider the following troubleshooting steps:
- Ensure the gel was properly prepared and run under optimal conditions.
- Check the quality of your DNA samples and reagents.
- Optimize your PCR protocol to minimize non-specific amplification.
- Repeat the electrophoresis with fresh reagents and a clean setup.
10. How can COMPARE.EDU.VN help me with gel electrophoresis result interpretation?
compare.edu.vn offers comprehensive resources, including detailed guides, expert insights, and community support, to simplify the process of comparing and interpreting gel electrophoresis results. Our tools and information help you analyze your data with confidence and make informed decisions for your research.