Can Blast Compare Amino Acids effectively? Yes, the Basic Local Alignment Search Tool (BLAST) is specifically designed for comparing biological sequences, including amino acid sequences, and COMPARE.EDU.VN offers extensive resources to understand and utilize this powerful tool. This article dives into the capabilities, applications, and nuances of using BLAST for amino acid sequence comparison, providing you with the knowledge to make informed decisions and perform effective sequence analysis.
1. What Is BLAST and How Does It Work?
BLAST, or Basic Local Alignment Search Tool, is an algorithm used to compare biological sequences. According to the National Center for Biotechnology Information (NCBI), BLAST finds regions of local similarity between sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches. BLAST can be used to infer functional and evolutionary relationships between sequences as well as help identify members of gene families.
1.1. Core Functionality of BLAST
BLAST’s primary function is to identify similar sequences within a vast database by comparing a query sequence against a collection of known sequences. This process involves aligning the query sequence with each sequence in the database, identifying regions of similarity, and assigning a score based on the quality of the alignment.
1.2. Types of BLAST Algorithms
Different BLAST algorithms are optimized for specific types of sequence comparisons:
- BLASTP: Compares an amino acid query sequence against a protein sequence database.
- BLASTN: Compares a nucleotide query sequence against a nucleotide sequence database.
- BLASTX: Compares a translated nucleotide query sequence against a protein sequence database.
- TBLASTN: Compares a protein query sequence against a translated nucleotide sequence database.
- TBLASTX: Compares a translated nucleotide query sequence against a translated nucleotide sequence database.
Each algorithm is tailored to handle specific types of input and database sequences, ensuring efficient and accurate comparisons.
1.3. Key Parameters in BLAST
Several parameters influence the sensitivity and specificity of BLAST searches:
- E-value (Expect value): Represents the number of alignments with scores equivalent to or better than the alignment score that are expected to occur by chance in a database search. Lower E-values indicate more significant alignments.
- Score: A numerical value reflecting the quality of the alignment between the query sequence and a database sequence. Higher scores indicate better alignments.
- Word size: The length of the initial seed words used to identify potential matches between the query sequence and database sequences. Larger word sizes result in faster searches but may miss weaker similarities.
- Gap penalties: Penalties applied for introducing gaps (insertions or deletions) in the alignment. Different gap penalties can affect the alignment score and the identification of homologous sequences.
2. Why Use BLAST for Amino Acid Sequence Comparison?
BLAST offers several advantages for comparing amino acid sequences, making it an indispensable tool in bioinformatics.
2.1. Identifying Homologous Proteins
BLAST is invaluable for identifying homologous proteins, which share a common evolutionary origin and often have similar functions. By comparing an unknown protein sequence against a database of known sequences, BLAST can reveal potential homologs, providing insights into the protein’s function and evolutionary history.
2.2. Predicting Protein Function
The identification of homologous proteins can aid in predicting the function of an unknown protein. If a query protein sequence shows significant similarity to a protein with a well-characterized function, it is likely that the query protein performs a similar function.
2.3. Analyzing Protein Structure
BLAST can be used to identify proteins with similar sequences and known structures. By aligning the query sequence with the sequences of proteins with known structures, researchers can generate homology models, which provide valuable insights into the three-dimensional structure of the query protein.
2.4. Discovering Conserved Domains and Motifs
BLAST can help identify conserved domains and motifs within protein sequences. These conserved regions often correspond to functionally important sites, such as catalytic domains or binding sites. The identification of conserved domains and motifs can provide clues about the protein’s function and mechanism of action.
3. How to Use BLAST for Amino Acid Comparison: A Step-by-Step Guide
Using BLAST for amino acid comparison involves several key steps, each requiring careful attention to detail.
3.1. Accessing BLAST
BLAST is freely available through the National Center for Biotechnology Information (NCBI) website. You can access BLAST via the following URL: https://blast.ncbi.nlm.nih.gov/Blast.cgi.
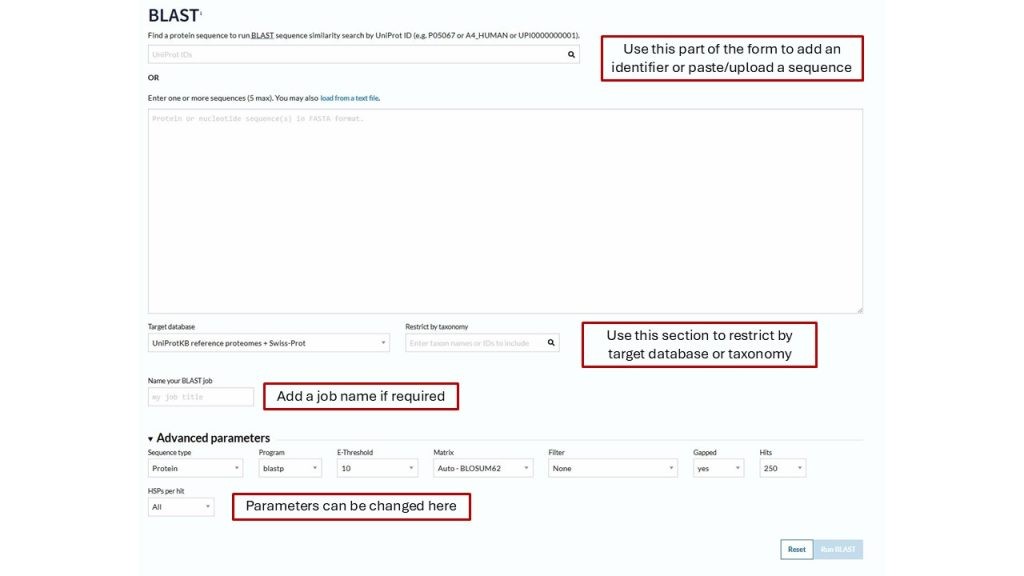
3.2. Inputting the Query Sequence
The first step is to input the query sequence, which is the amino acid sequence you want to compare against the database. You can enter the sequence in FASTA format or as a raw sequence. Ensure that the sequence is accurate and free of errors to obtain reliable results.
3.3. Selecting the Appropriate BLAST Program
Choose the appropriate BLAST program based on the type of query sequence and the database you want to search. For amino acid sequence comparison, select BLASTP, which is specifically designed for comparing protein sequences against protein databases.
3.4. Choosing the Database
Select the appropriate database to search against. NCBI offers a variety of databases, including:
- nr (Non-redundant protein sequences): A comprehensive database containing protein sequences from various sources.
- swissprot: A curated protein sequence database providing high-quality annotations.
- pdb (Protein Data Bank): A database of experimentally determined protein structures.
The choice of database depends on the specific research question and the type of sequences you are interested in comparing.
3.5. Setting Parameters
Adjust the BLAST parameters to optimize the search for your specific needs. Key parameters to consider include:
- E-value: Set an appropriate E-value threshold to filter out insignificant matches. A lower E-value (e.g., 1e-5 or 1e-10) will result in more stringent results.
- Word size: Adjust the word size to balance sensitivity and speed. Smaller word sizes are more sensitive but slower, while larger word sizes are faster but less sensitive.
- Gap penalties: Modify the gap penalties to fine-tune the alignment algorithm.
3.6. Running the BLAST Search
After setting the parameters, run the BLAST search. The time required for the search depends on the size of the query sequence and the database.
3.7. Analyzing the Results
Analyze the BLAST results to identify significant matches. Key information to consider includes:
- Score: The alignment score, which reflects the quality of the alignment.
- E-value: The expect value, which indicates the statistical significance of the match.
- Identity: The percentage of identical amino acids between the query sequence and the database sequence.
- Query coverage: The percentage of the query sequence that is covered by the alignment.
Examine the alignments to identify conserved regions and potential functional domains.
4. Interpreting BLAST Results for Amino Acid Sequences
Interpreting BLAST results requires a thorough understanding of the various parameters and statistical measures provided in the output.
4.1. Understanding E-values and Significance
The E-value is a critical parameter for assessing the significance of BLAST hits. It represents the probability of finding a match with the observed score by chance. Lower E-values indicate more significant matches. For example, an E-value of 1e-5 means that there is only a 0.00001 probability of finding a match with that score by chance.
4.2. Evaluating Alignment Scores
The alignment score reflects the quality of the alignment between the query sequence and the database sequence. Higher scores indicate better alignments. The score is calculated based on the substitution matrix, which assigns scores to different amino acid pairings.
4.3. Assessing Percentage Identity and Similarity
The percentage identity indicates the proportion of identical amino acids between the query sequence and the database sequence. The percentage similarity considers both identical and similar amino acids. High percentage identity and similarity suggest a close evolutionary relationship between the sequences.
4.4. Identifying Conserved Domains and Motifs from BLAST Hits
BLAST hits can reveal conserved domains and motifs within the query sequence. Conserved domains are regions of the protein sequence that are highly conserved across different species, indicating their functional importance. Motifs are short, conserved sequence patterns that often correspond to specific functional sites.
5. Advanced BLAST Techniques for Amino Acid Comparison
Advanced BLAST techniques can enhance the accuracy and sensitivity of amino acid sequence comparisons.
5.1. PSI-BLAST (Position-Specific Iterated BLAST)
PSI-BLAST is an iterative search algorithm that improves the sensitivity of BLAST searches by constructing a position-specific scoring matrix (PSSM) from the initial BLAST hits. The PSSM is then used to search the database again, identifying more distant homologs that may have been missed in the initial search.
5.2. DELTA-BLAST (Domain Enhanced Lookup Time Accelerated BLAST)
DELTA-BLAST is another advanced BLAST algorithm that uses precomputed domain information to improve the accuracy of sequence comparisons. DELTA-BLAST identifies conserved domains within the query sequence and uses this information to guide the search for homologous sequences.
5.3. Using Custom Databases
Researchers can create custom databases to focus their BLAST searches on specific sets of sequences. This can be useful for identifying homologs within a particular taxonomic group or for analyzing sequences from a specific experiment.
5.4. Filtering and Masking Sequences
Filtering and masking sequences can improve the accuracy of BLAST searches by removing regions that may lead to false positives. Filtering removes low-complexity regions, while masking replaces repetitive sequences with ambiguous characters.
6. Common Issues and Troubleshooting in BLAST
Despite its power and versatility, BLAST can sometimes produce unexpected or misleading results.
6.1. Dealing with Low-Complexity Regions
Low-complexity regions can lead to spurious hits in BLAST searches. These regions consist of repetitive or biased amino acid compositions that can match many unrelated sequences. Filtering low-complexity regions can reduce the number of false positives.
6.2. Addressing Database Redundancy
Database redundancy can complicate the interpretation of BLAST results. Highly similar sequences can appear multiple times in the database, leading to multiple hits for the same protein. Clustering sequences to reduce redundancy can simplify the analysis.
6.3. Handling Short or Fragmented Sequences
Short or fragmented sequences can be challenging to analyze using BLAST. These sequences may not contain enough information to identify significant homologs. Using more sensitive search parameters or combining multiple fragments can improve the results.
6.4. Avoiding Over-Interpretation of Results
It is important to avoid over-interpreting BLAST results. The identification of a homologous sequence does not necessarily imply that the query protein has the same function as the database protein. Additional experimental evidence is needed to confirm functional predictions.
7. Real-World Applications of BLAST in Amino Acid Research
BLAST plays a crucial role in various fields of biological research, enabling scientists to make significant discoveries.
7.1. Drug Discovery and Development
BLAST is used in drug discovery to identify potential drug targets. By comparing the sequences of disease-related proteins with the sequences of known drug targets, researchers can identify proteins that may be susceptible to drug intervention.
7.2. Personalized Medicine
BLAST is used in personalized medicine to identify genetic variations that may affect an individual’s response to drugs. By comparing an individual’s DNA sequence with reference sequences, researchers can identify variations that may alter the structure or function of drug-metabolizing enzymes.
7.3. Agricultural Biotechnology
BLAST is used in agricultural biotechnology to identify genes that may improve crop yields or resistance to pests and diseases. By comparing the sequences of plant genes with the sequences of genes from other organisms, researchers can identify genes that may confer desirable traits.
7.4. Environmental Science
BLAST is used in environmental science to identify microorganisms that may be useful for bioremediation or other environmental applications. By comparing the sequences of microbial genes with the sequences of genes from known organisms, researchers can identify microorganisms that may have the ability to degrade pollutants or produce valuable products.
8. How COMPARE.EDU.VN Enhances Your Understanding of BLAST
COMPARE.EDU.VN serves as a comprehensive resource for understanding and utilizing BLAST effectively.
8.1. Providing Detailed Tutorials and Guides
COMPARE.EDU.VN offers detailed tutorials and guides on using BLAST for amino acid sequence comparison. These resources cover the basics of BLAST, as well as advanced techniques and troubleshooting tips.
8.2. Offering Comparative Analyses of Different BLAST Algorithms
COMPARE.EDU.VN provides comparative analyses of different BLAST algorithms, helping users choose the most appropriate algorithm for their specific needs. These analyses compare the sensitivity, specificity, and speed of different algorithms.
8.3. Presenting Case Studies and Real-World Examples
COMPARE.EDU.VN presents case studies and real-world examples of BLAST applications, illustrating how BLAST can be used to solve real-world problems in biological research.
8.4. Facilitating Community Discussions and Knowledge Sharing
COMPARE.EDU.VN facilitates community discussions and knowledge sharing, allowing users to ask questions, share tips, and learn from each other’s experiences.
9. The Future of BLAST and Amino Acid Sequence Comparison
The field of amino acid sequence comparison is constantly evolving, with new algorithms and technologies emerging.
9.1. Integration with Machine Learning and AI
The integration of machine learning and artificial intelligence is transforming the field of amino acid sequence comparison. Machine learning algorithms can be used to improve the accuracy of sequence alignments, predict protein function, and identify novel drug targets.
9.2. Development of More Sensitive and Accurate Algorithms
Researchers are constantly developing more sensitive and accurate algorithms for amino acid sequence comparison. These algorithms incorporate new statistical models, alignment techniques, and database search strategies.
9.3. Expansion of Sequence Databases
The expansion of sequence databases is providing researchers with access to an ever-growing collection of biological sequences. This is enabling the discovery of new genes, proteins, and evolutionary relationships.
9.4. Improved Visualization and Interpretation Tools
Improved visualization and interpretation tools are making it easier for researchers to analyze and interpret BLAST results. These tools provide interactive displays of sequence alignments, functional annotations, and evolutionary relationships.
10. Conclusion: Mastering Amino Acid Comparison with BLAST
BLAST is a powerful tool for comparing amino acid sequences, enabling researchers to identify homologous proteins, predict protein function, analyze protein structure, and discover conserved domains and motifs. By mastering the use of BLAST and understanding its underlying principles, researchers can make significant contributions to the fields of biology, medicine, and biotechnology. Remember to leverage resources like COMPARE.EDU.VN to enhance your understanding and skills in sequence analysis.
Ready to take your understanding of amino acid comparison to the next level? Visit COMPARE.EDU.VN today to explore comprehensive tutorials, comparative analyses, and real-world examples that will empower you to make informed decisions and drive your research forward. For further inquiries, contact us at 333 Comparison Plaza, Choice City, CA 90210, United States, or reach out via WhatsApp at +1 (626) 555-9090. Your journey to mastering sequence analysis starts here!
Frequently Asked Questions (FAQs)
1. What is the primary purpose of using BLAST for amino acid sequence comparison?
The primary purpose is to identify homologous proteins, predict protein function, analyze protein structure, and discover conserved domains and motifs.
2. How does the E-value affect the interpretation of BLAST results?
A lower E-value indicates a more significant match, suggesting a lower probability that the match occurred by chance.
3. What is PSI-BLAST, and how does it improve sequence comparison?
PSI-BLAST is an iterative search algorithm that improves sensitivity by constructing a position-specific scoring matrix (PSSM) from initial hits to find more distant homologs.
4. Why is it important to filter low-complexity regions in BLAST searches?
Filtering low-complexity regions reduces spurious hits by removing repetitive or biased amino acid compositions that can match unrelated sequences.
5. How can COMPARE.EDU.VN help in understanding and using BLAST?
compare.edu.vn provides detailed tutorials, comparative analyses, case studies, and facilitates community discussions for better understanding and utilization of BLAST.
6. What are some real-world applications of using BLAST in amino acid research?
Real-world applications include drug discovery, personalized medicine, agricultural biotechnology, and environmental science.
7. What is the significance of the percentage identity and similarity in BLAST results?
High percentage identity and similarity suggest a close evolutionary relationship between the query and database sequences.
8. How do advanced BLAST techniques like DELTA-BLAST enhance sequence comparisons?
DELTA-BLAST uses precomputed domain information to improve the accuracy of sequence comparisons, identifying conserved domains within the query sequence.
9. What are some potential issues when using BLAST with short or fragmented sequences?
Short or fragmented sequences may not contain enough information to identify significant homologs, requiring more sensitive search parameters or combining multiple fragments.
10. How is the integration of machine learning and AI transforming amino acid sequence comparison?
Machine learning algorithms improve the accuracy of sequence alignments, predict protein function, and identify novel drug targets, enhancing the overall process of sequence comparison.