Vibrio is recognized as a highly diverse genus within the Gammaproteobacteria class, predominantly found in marine environments globally. Vibrio diabolicus and V. antiquarius were initially identified from deep-sea hydrothermal vents in the East Pacific Rise. Interestingly, these species exhibit a close phylogenetic relationship to members of the Harveyi clade, such as V. alginolyticus and V. parahaemolyticus, which are frequently isolated from coastal ecosystems. This research details the discovery and initial genome sequencing of a novel isolate, Vibrio sp. 939, cultured from Pacific oysters (Crassostrea gigas). Questions arising from the classification of Vibrio sp. 939 prompted a comprehensive, genome-scale taxonomic analysis focusing on the Harveyi clade and the concept of Comparative Synonymy within bacterial species. A robust phylogenetic analysis, incorporating 49 genomes and based on 1,109 conserved coding sequences, alongside a comparison of Average Nucleotide Identity (ANI) values, conclusively demonstrated a clear case of comparative synonymy. Our findings reveal that Vibrio sp. 939, V. diabolicus strains Art-Gut C1 and CNCM I-1629, V. antiquarius EX25, and notably, four V. alginolyticus strains (E0666, FF273, TS13, and V2) are indeed synonymous. This discovery significantly expands the definition of V. diabolicus as a species, providing six additional genomes for further comparative genomic studies and highlighting the importance of comparative synonymy in bacterial taxonomy. The expanded species exhibits a global distribution, evidenced by its isolation from diverse sources including horse mackerel, seawater, sediment, dentex, oyster, artemia, and polychaete, across a broad geographical range encompassing China, India, Greece, the United States, the East Pacific Rise, and Chile. Subsequent comparative genomic analysis of this newly defined eight-genome subclade revealed substantial individual genome plasticity and a rich set of genes associated with virulence and defense mechanisms. These findings necessitate a significant revision in our understanding of V. diabolicus and V. antiquarius, species previously considered distinct. This initial exploration of the expanded V. diabolicus subclade indicates that its distribution and diversity are comparable to other Harveyi clade species, known for their widespread presence and remarkable diversity.
Introduction
The genus Vibrio stands as a diverse collection of Gram-negative bacteria (Thompson et al., 2004), encompassing approximately 139 recognized species1. Since Vibrio cholerae was first isolated by Filippo Pacini in 1854 (Pacini, 1854), this genus has been the subject of extensive research due to the ease of culturing many species and the fact that at least twelve are pathogenic to humans (Chakraborty et al., 1997). Notably, both V. cholerae and V. parahaemolyticus have been implicated in pandemic outbreaks (Karaolis et al., 1995; Matsumoto et al., 2000), while V. vulnificus is recognized for its capacity to cause primary sepsis, particularly in immunocompromised individuals (Strom and Paranjpye, 2000). The genus also includes species pathogenic to commercially important fisheries and foundational marine organisms. For example, V. harveyi and V. anguillarum are pathogens affecting penaeid shrimp and fish, respectively (Austin and Zhang, 2006; Frans et al., 2011), and V. coralliilyticus and V. shiloi are known coral pathogens (Rosenberg et al., 2007). Despite the pathogenic potential of some species, the genus is considered conditionally rare (Shade et al., 2014), with many species being benign (Thompson and Polz, 2006; Thompson et al., 2006).
Vibrio species exhibit a broad geographic distribution, commonly found in riverine, estuarine, and coastal marine ecosystems worldwide (Thompson et al., 2004). Sea surface temperature often acts as a limiting factor, confining growth to warmer latitudes due to the mesophilic nature of many species (Takemura et al., 2014). However, two Vibrio species were initially isolated from environments distinctly removed from coastal ecosystems and direct human interaction. V. diabolicus (strain CNCM I-1629) was first isolated from a polychaete associated with a deep-sea hydrothermal vent in the East Pacific Rise (Raguénès et al., 1997, where temperatures are conducive to mesophilic growth. Similarly, V. antiquarius (strain EX25) was first isolated from seawater near a sulfide chimney in the East Pacific Rise (Hasan et al., 2015).
Previous studies have suggested that V. diabolicus CNCM I-1629 and V. antiquarius EX25 possess specific genes that facilitate survival and persistence in hydrothermal vent ecosystems. For instance, V. diabolicus CNCM I-1629 produces an acidic exopolysaccharide (EPS) thought to provide resistance to high concentrations of metallic sulfides (e.g., pyrite, chalcopyrite, and sphalerite) found in sulfide chimneys (Raguénès et al., 1997; Goudenège et al., 2014). The genome of V. antiquarius EX25 encodes an alkyl hydroperoxide reductase, proposed to neutralize endogenous hydrogen peroxide in the deep-sea environment (Hasan et al., 2015), as this enzyme is also found in the deep-sea tubeworm endosymbiont Riftia pachyptila (Markert et al., 2007). Intriguingly, both species also carry hemolysins and type III secretion systems (T3SS), typically considered virulence-associated factors in pathogenic Vibrio species (Goudenège et al., 2014; Hasan et al., 2015). The presence of these virulence factors in deep-sea species has been interpreted as evidence that secreted toxins are evolutionarily ancient features potentially playing a broader role in environmental adaptation (Hasan et al., 2015).
The distribution, diversity, and potential virulence of these deep-sea Vibrio species remain largely unexplored. It is plausible that both V. diabolicus and V. antiquarius exhibit a cosmopolitan distribution, a broader range that would align with the adaptability of closely related Vibrio species, such as V. alginolyticus and V. parahaemolyticus. These species are characterized as opportunitrophs, capable of utilizing resources that are both spatially and temporally limited (Polz et al., 2006). Indeed, the detection of V. antiquarius EX25 open reading frames (ORFs) in 89 shotgun metagenomic datasets (Hasan et al., 2015) and the recent isolation of twelve V. diabolicus isolates in a mid-Atlantic estuary (Klein et al., 2014) support the hypothesis of a wider distribution for these deep-sea Vibrio species.
In the course of an extensive, multifaceted study on V. parahaemolyticus (Johnson et al., 2012; Paranjpye et al., 2012; Turner et al., 2013, 2016), Vibrio sp. 939 was isolated from Pacific oysters (Crassostrea gigas) harvested from Puget Sound in the Pacific Northwest of the United States. This paper presents the discovery and draft genome sequencing of Vibrio sp. 939 and details the investigation into its identity through a comprehensive taxonomic analysis of 49 Harveyi clade genomes. The findings unequivocally demonstrate that V. antiquarius and V. diabolicus represent the same species, and we propose V. diabolicus as the senior synonym. Furthermore, the data reveal that four strains previously misidentified as V. alginolyticus (E0666, FF273, TS13, and V2) are also synonyms of V. diabolicus. Subsequent comparative genomic analysis addresses critical questions about the distribution and diversity of this expanded species.
Materials and Methods
Sample Collection
Vibrio sp. 939 was isolated from Pacific oysters (C. gigas) collected from Hood Canal, part of Puget Sound in Washington State, USA. This isolate was among hundreds collected by the NOAA Northwest Fisheries Science Center (Seattle, WA, USA) during a comprehensive V. parahaemolyticus study (Johnson et al., 2012; Paranjpye et al., 2012; Turner et al., 2013, 2016). Briefly, oysters were scrubbed, shucked, and homogenized. Presumptive Vibrio species were isolated by direct plating on thiosulfate-citrate-bile salts-sucrose (TCBS) agar (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and incubated overnight at 30°C. Vibrio sp. 939 was initially presumed to be V. parahaemolyticus based on the detection of the thermolabile hemolysin (tlh) gene (Bej et al., 1999. However, it was excluded from a prior V. parahaemolyticus multilocus sequence typing (MLST) study (Turner et al., 2013) due to amplification issues with many MLST loci. This preliminary identification was also questioned by recent studies showing that V. parahaemolyticus-specific tlh primers could also amplify sequence variants of the tlh gene in closely related species (Klein et al., 2014; Yáñez et al., 2015).
Culture Conditions
Starting from a frozen cell culture (25% glycerol, v/v, -80°C), Vibrio sp. 939 was streaked for isolation on lysogeny broth (LB) (Fisher Scientific, Fair Lawn, NJ, USA) supplemented with 1.5% Bacto agar (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and grown for 18 hours at 30°C. A single isolated colony was then transferred to 5 mL of LB in a 15 mL BD Falcon tube (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and cultured for 18 hours at 30°C with shaking (120 rpm) using an Excella E24 shaking incubator (Eppendorf, Hamburg, Germany).
DNA Isolation
Bacteria from the overnight culture (1 mL) were pelleted by centrifugation (9,400 × g, 5 min) in an Eppendorf 5424E centrifuge (Hamburg, Germany) and washed twice with an equal volume of phosphate-buffered saline (PBS). DNA was extracted from the pelleted cells using a ChargeSwitch gDNA Mini Bacteria Kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. DNA was quantified and quality assessed (A260/A280) using a BioPhotometer D30 (Eppendorf, Hamburg, Germany) and stored at -20°C.
Genome Sequencing
Genomic DNA sequencing was performed using an Illumina MiSeq instrument at the New York University Genome Technology Center, employing paired-end chemistry (2 × 300 bp). The sequencing library was prepared using the PCR-free version of a KAPA DNA Library Preparation Kit (Kapa Biosystems, Wilmington, MA, USA). Overlapping paired reads were merged using FLASH version 1.2.11 (Magoc and Salzberg, 2011). Adapter sequences and low-quality bases were trimmed from the merged reads using Trim Galore! version 0.4.42, a wrapper script for Cutadapt (Martin, 2011) and FastQC (Andrews, 2010). The optimal k-mer size was estimated with KmerGenie version 1.7 (Chikhi and Medvedev, 2014), and de novo genome assembly was performed using Velvet version 1.2.10 (Zerbino and Birney, 2008). Initial genome annotation and inspection were conducted using the web-based RAST annotation service and SEED Viewer (Aziz et al., 2008; Overbeek et al., 2014). Final annotation was completed using the National Center for Biotechnology Information’s (NCBI) Prokaryotic Genome Annotation Pipeline (PGAP) (Klimke et al., 2009).
Phylogenetics
To determine the phylogenetic relationship between Vibrio sp. 939 and 48 closely related Harveyi clade species (including V. diabolicus, V. antiquarius, V. parahaemolyticus, V. alginolyticus, and V. harveyi) (Supplementary Table S1), a maximum-likelihood tree was constructed. This tree was based on a set of single-copy homologs present in all 49 Harveyi clade genomes, downloaded from NCBI. Single-copy homologous genes were clustered using get_homologues (options -M -t 49 -r EX25.gbk -e) (Contreras-Moreira and Vinuesa, 2013). The OrthoMCL algorithm (Li et al., 2003) was used for homolog search with a default E-value cutoff (1e-05). Clustering was limited to single-copy homologs present in all 49 genomes, using the closed genome of V. antiquarius EX25 as a reference, and clusters containing in-paralogs were excluded. Homologous clusters were individually aligned with MUSCLE version 3.6 (Edgar, 2004), and alignments were trimmed to the length of the shortest sequence using trimAl version 1.4.15 (Capella-Gutierrez et al., 2009). Trimmed alignments were filtered by length (250 bp cutoff), concatenated using the fasta manipulation tool in Galaxy (Giardine et al., 2005), and a maximum-likelihood tree was constructed with IQ-TREE version 1.5.5 (Nguyen et al., 2014) with 1,000 ultrafast bootstraps (Minh et al., 2013), employing the best-fit model (GTR+I+G4) as determined by ModelFinder (Kalyaanamoorthy et al., 2017). The phylogenetic tree was annotated using FigTree version 1.4.33.
Phylogenetic Network
To visualize potential conflicting phylogenetic signals resulting from reticulate evolutionary processes, a Neighbor-Net phylogenetic network (Bryant and Moulton, 2004) was constructed for the concatenated multilocus alignment (described above). SplitsTree4 version 4.14.4 (Huson and Bryant, 2006) was used with default settings and 1,000 bootstraps.
Genome Similarity
Genome similarity between V. diabolicus Art-Gut C1 and CNCM I-1629, Vibrio sp. 939, V. antiquarius EX25, and V. alginolyticus 12G01, 40B, E0666, FF273, K01M1, NBRC 15630, TS13, and V2 was assessed using all-versus-all alignment with JSpeciesWS (Richter et al., 2016). JSpeciesWS, a web server implementation of the JSpecies Taxonomic Thresholds Program (Richter and Rosselló-Móra, 2009), calculates Average Nucleotide Identity (ANI) between a query and reference genome, with a 95–96% similarity threshold for prokaryotic species identification. To further illustrate sequence similarity within the eight V. diabolicus subclade genomes (V. diabolicus Art-Gut C1 and CNCM I-1629, Vibrio sp. 939, V. antiquarius EX25, and V. alginolyticus E0666, FF273, TS13, and V2), a circular blast map was generated using the BLAST Ring Image Generator (BRIG) (Alikhan et al., 2011). Identity thresholds were set at 92 and 96%, with V. antiquarius EX25 as the reference genome. V. alginolyticus NBRC 15630 was included for comparison. Genome attributes (size, %GC content, total genes, protein-coding genes, RNA genes) were also calculated to compare these eight genomes.
Pangenome Analysis
To evaluate the diversity of the V. diabolicus subclade, pangenome analysis was conducted using two methods. First, get_homologues was used with option t = 0 to report all homologous gene clusters. The parse_pangenome_matrix.pl script was used to identify core genes (present in all eight strains) and accessory genes (present in fewer than eight strains). Recognizing that get_homologues excludes clusters with fewer than three sequences and non-coding sequences, a second pangenome analysis was performed using Panseq (Laing et al., 2010) with default parameters. Panseq uses all-versus-all alignment of genomic regions to estimate the core, accessory, and pangenome. For this analysis, the core genome was defined as genomic regions present in all eight genomes. Panseq’s Novel Region Finder was used to identify singletons (genomic regions unique to one genome), filtered by length (10 Kb cutoff) to detect probable genomic islands (GIs). These GIs were then inspected in the web-based SEED Viewer (blastn E-value cutoff 1e-05) (Overbeek et al., 2014) for habitat-specific functions.
Subclade-Associated Genes
Output from pangenome analysis was parsed using parse_pangenome_matrix.pl to identify genes ubiquitous in the V. diabolicus subclade (N = 8 strains) but absent from the other 41 strains. Subclade-associated genes and their protein products were individually aligned with MUSCLE version 3.6 (Edgar, 2004), and average sequence identity for each alignment was calculated using trimAl version 1.4.15 (Capella-Gutierrez et al., 2009).
Metagenome Survey
To investigate the presence of V. diabolicus in public metagenomic data, protein sequences from the most conserved non-hypothetical subclade-associated genes were queried against the NCBI Metagenome Protein Database (env_nr) using default blastp parameters. Significant hits were defined as having sequence identity comparable to subclade protein alignments (greater than 95%).
Potential Virulence
The prevalence and diversity of virulence and defense-related genes were investigated using SEED subsystem feature counts in the web-based SEED Viewer. Eight genomes of the V. diabolicus subclade were examined for proteins with significant matches (blastp, E-value cutoff 1e-05) to five subsystems: (1) Adhesion, (2) Toxins and Superantigens, (3) Bacteriocins and Ribosomally Synthesized Antibacterial Peptides, (4) Resistance to Antibiotics and Toxic Compounds, and (5) Invasion and Intracellular Resistance. Specifically, β-lactamase proteins were aligned with MUSCLE version 3.6 (Edgar, 2004), trimmed with trimAl version 1.4.15 (Capella-Gutierrez et al., 2009), and a maximum-likelihood tree was constructed with IQ-TREE version 1.5.5 (Nguyen et al., 2014) with 1,000 ultrafast bootstraps (Minh et al., 2013), using the best-fit model (WAG) determined by ModelFinder (Kalyaanamoorthy et al., 2017). β-lactamase proteins (present in all genomes) and fosfomycin resistance proteins (V. alginolyticus TS13 and V2) were searched against the Antibiotic Resistance Genes Database (ARDB) using default parameters (blastp, E-value cutoff 1e-05) (Liu and Pop, 2009).
Antibiotic Susceptibility
β-lactamase production was tested using Cefinase disks (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) following manufacturer instructions. Susceptibility to β-lactam antibiotics was determined as previously described (Letchumanan et al., 2015). V. antiquarius 939 was cultured overnight on Mueller-Hinton agar plates (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) (supplemented with 1.5% NaCl) at 37°C. Overnight growth was resuspended in filter-sterilized 0.85% NaCl solution, normalized to a 0.5 McFarland standard, and fresh Mueller-Hinton agar plates were seeded with a bacterial lawn. Penicillin (10 units), ampicillin (10 μg), cephalothin (30 μg), and carbenicillin (100 μg) antibiotic disks (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) were placed on the lawn, and plates were incubated for 18 hours at 37°C. Zones of inhibition were measured and interpreted according to Clinical and Laboratory Standards Institute (CLSI) guidelines (Clinical and Laboratory Standards Institute [CLSI], 2010). The pandemic type strain of V. parahaemolyticus (RIMD2210633) (Nasu et al., 2000) was tested for comparison.
Results
Genome Sequencing
The draft genome of Vibrio sp. 939 consisted of 46 contigs, totaling 5,430,661 bp with 44.6% GC content. The N50 was 855,346 bp, and contig lengths ranged from 570 to 1,035,854 bp. PGAP annotation identified 5,049 genes, including 4,876 proteins, 112 RNAs (13 rRNA, 95 tRNA, and 4 other RNA), and 61 pseudogenes. Based on initial SEED Viewer evaluation, the whole-genome shotgun project was deposited as V. antiquarius 939 at DDBJ/ENA/GenBank under accession number AOJB00000000.
Phylogenetic Tree
Homologous gene search across 49 genomes yielded 1,125 gene clusters. After length filtering, 1,109 single-copy homologs remained. Concatenation produced a multilocus alignment of 1,034,083 bp, with 595,014 constant sites and 404,946 parsimony informative sites, and 217,816 distinct site patterns. The best-fit model was GTR+I+G4.
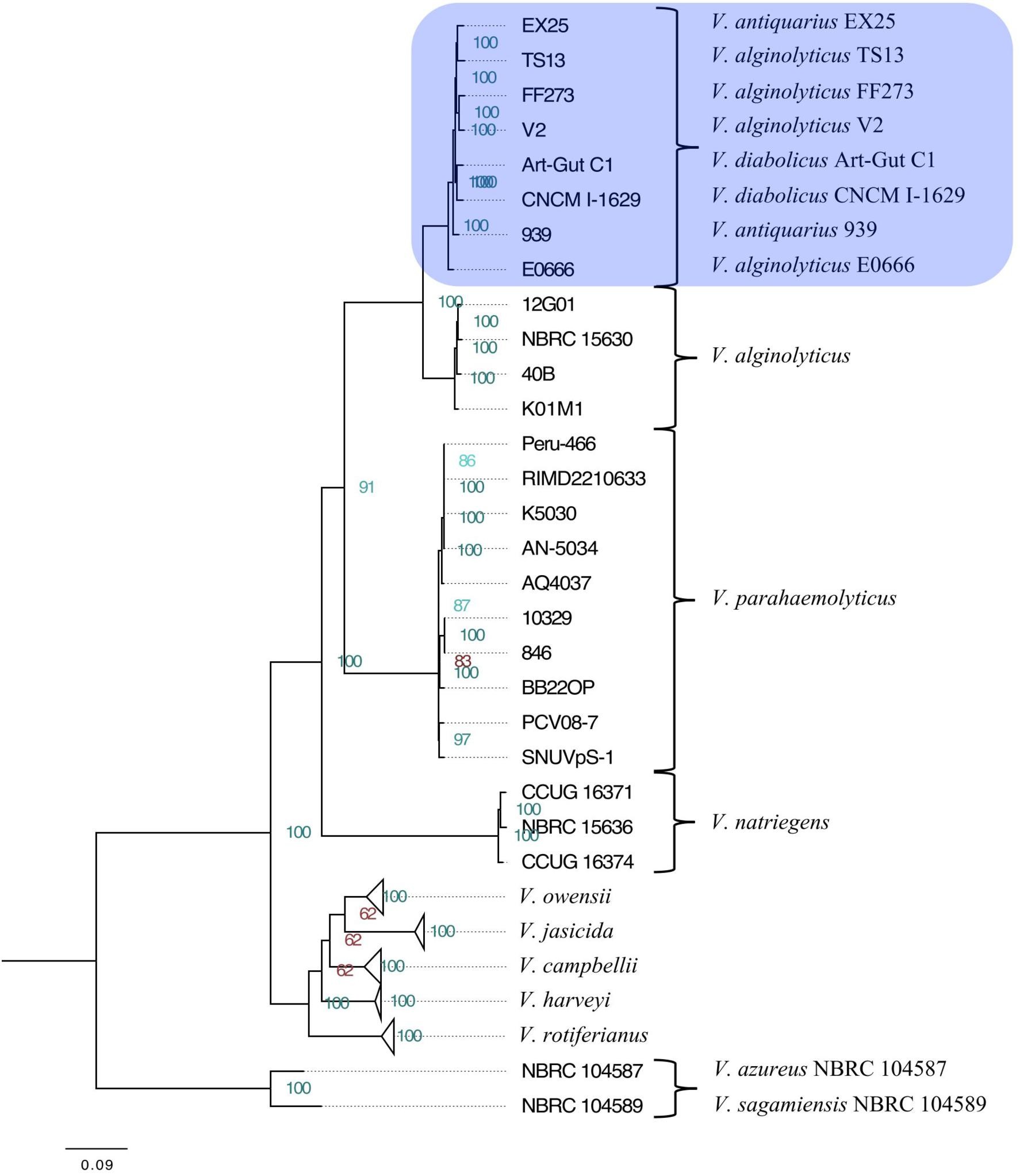
The maximum-likelihood phylogeny resolved the Harveyi clade into eleven subclades, including V. alginolyticus, V. azureus, V. campbellii, V. harveyi, V. jasicida, V. natriegens, V. owensii, V. parahaemolyticus, V. rotiferianus, V. sagamiensis, and a mixed subclade. This mixed subclade comprised two V. diabolicus strains (Art-Gut C1 and CNCM I-1629), two V. antiquarius strains (939 and EX25), and four V. alginolyticus strains (E0666, FF273, TS13, and V2) (Figure 1). The phylogeny was consistent with previous whole-genome Harveyi clade phylogenies, but uniquely resolved non-core Harveyi subclades. The novel, well-supported V. diabolicus subclade was particularly notable.
Phylogenetic Network
The Neighbor-Net network, using the same 1,109 multilocus alignment, mirrored the phylogenetic tree, showing strong support for the V. diabolicus subclade. Reticulation was more evident between the V. diabolicus and V. alginolyticus subclades, suggesting potential recombination. However, bootstrap analysis strongly supported the distinct V. diabolicus subclade.
Genome Similarity
Pairwise ANI values (Table 1) showed that the eight genomes in the V. diabolicus subclade shared greater than 97% ANI. In contrast, they shared less than 92% ANI with representative V. alginolyticus genomes.
The circular blastn map (Figure 3) visually confirmed high synonymy among the V. diabolicus subclade genomes, including V. diabolicus, V. antiquarius, and the four V. alginolyticus strains. Non-synonymy with V. alginolyticus NBRC 15630 was also evident. Hypervariable regions included a capsular polysaccharide region, a super integron, a type VI secretion system (T6SS), and a glucuronic acid utilization cluster.
Isolates in the V. diabolicus subclade originated from diverse sources and global locations. Genome sizes ranged from 5,048,917 to 5,430,661 bp, gene counts from 4,572 to 5,251, and GC content from 44.6 to 44.9% (Table 2).
Genome Diversity
Pangenome analysis using get_homologues revealed 4,854 total and 3,422 core genes in the V. diabolicus subclade, with accessory genes comprising ∼29.5% of the pangenome. Panseq analysis indicated a 7,856,358 bp pangenome, with a 4,605,287 bp core genome (∼58.6%) and a 3,251,071 bp accessory genome (∼41.4%).
Panseq Novel Region Finder identified 2,295,535 bp of novel genomic regions within the accessory genome, with probable GIs (>10 Kb) prevalent in each genome (Figure 4). These GIs encoded diverse proteins, including hypothetical proteins, polysaccharide biosynthesis proteins, metabolic enzymes, transport proteins, phage-related proteins, and conjugative transfer proteins, but lacked obvious habitat-specific functions. A V. alginolyticus TS13 GI contained a widespread colonization island (WCI) with a tad locus. Alkyl hydroperoxide reductase was found in all eight V. diabolicus strains.
Subclade-Associated Genes
Five genes were ubiquitous in the V. diabolicus subclade but absent from other strains (Table 3). These single-copy, highly conserved genes included two encoding hypothetical proteins and three encoding a hydroxyectoine utilization dehydratase, an ornithine cyclodeaminase, and a phosphoesterase.
Metagenome Survey
Querying the NCBI Metagenome Protein Database with three subclade-associated coding sequences (hydroxyectoine utilization dehydratase, ornithine cyclodeaminase, and phosphoesterase) yielded non-significant hits. Sequence identity was below 95%, indicating these markers were not readily detected in environmental metagenomes at high similarity.
Potential Virulence
SEED subsystem feature comparison revealed a large repertoire of virulence and defense features, with nearly identical profiles across the V. diabolicus subclade genomes (Figure 5). Accessory colonization factor AcfA, colicin V production cluster features, and proteins homologous to a Mycobacterium virulence operon were consistently present. Toxins and Superantigens subsystem features varied but included ToxR, ToxS, Zonula occludens toxin (Zot), and accessory cholera enterotoxin (Ace).
Evaluation of Resistance to Antimicrobial Compounds subsystem features showed 74–79 features per genome, with a nearly identical resistome, including resistance to toxic metals/metalloids and antibiotics (fluoroquinolones, fosfomycin, tetracycline, penicillin), and 22–24 efflux pumps per genome (MATE and RND families).
Phylogenetic analysis of β-lactamase proteins revealed two distinct β-lactamases (Supplementary Figure S2), identified as class A and class C β-lactamases, associated with carbenicillin and cephalosporin resistance, respectively. FosA enzymes in V. alginolyticus TS13 and V2 were identified as glutathione transferases conferring fosfomycin resistance.
Antibiotic Susceptibility
A positive Cefinase test confirmed β-lactamase production in V. antiquarius 939 and V. parahaemolyticus RIMD2210633A. V. antiquarius 939 showed resistance to penicillin, ampicillin, and cephalothin, but susceptibility to carbenicillin. V. parahaemolyticus RIMD2210633 was resistant to penicillin and cephalothin, intermediately susceptible to ampicillin, and susceptible to carbenicillin.
Discussion
The taxonomy of the Vibrio genus has expanded rapidly, growing from five recognized species in the 1970s to approximately 139 today. This expansion is accompanied by ongoing taxonomic revisions, particularly within the Harveyi clade, where species boundaries have been challenging to define. Multilocus and genome-scale phylogenetics have significantly advanced our understanding of this clade. This study builds upon this progress by clarifying the relationships of deep-sea Harveyi clade species V. diabolicus and V. antiquarius.
The identity of Vibrio sp. 939 prompted genome sequencing and a taxonomic analysis of 49 Harveyi clade genomes. Initial RAST analysis suggested a close relationship to V. antiquarius, leading to its initial GenBank assignment. However, a 49-genome phylogeny based on 1,109 homologous genes placed the isolate within a monophyletic subclade, also including V. diabolicus, V. antiquarius, and four V. alginolyticus strains.
Soft species boundaries within the Harveyi clade may result from recombination, complicating phylogenetic signal. A phylogenetic network analysis revealed short branching between V. jasicida, V. harveyi, V. owensii, and V. campbellii, and between V. alginolyticus and V. diabolicus. Reticulation between V. alginolyticus and V. diabolicus suggests recombination may obscure phylogenetic signals, potentially explaining why the boundary between these species remained unclear. Despite this, network analysis subclades were well-supported and consistent with the maximum-likelihood phylogeny.
Applying the 95–96% ANI threshold for species delineation, the study found that V. diabolicus, V. antiquarius, and the four V. alginolyticus genomes shared >97% ANI, strongly indicating they belong to the same species. They shared <92% ANI with V. alginolyticus genomes, confirming their distinctiveness from this closest neighbor. Similar ANI thresholds have been used to establish comparative synonymy in other Vibrio species. Our ANI analysis firmly establishes that V. antiquarius strains and the four V. alginolyticus strains are comparative synonyms of V. diabolicus.
The diverse origins of the eight V. diabolicus strains suggest broad physiological capabilities. Genomic variability, reflected in a large accessory genome and strain-specific GIs, may facilitate adaptation to diverse habitats. For example, the tad locus-encoded Flp pilus could promote colonization. However, the species appears conditionally rare, as species-associated genetic markers were not readily detected in metagenomic databases.
The comparative synonymy of V. antiquarius with V. diabolicus significantly revises the taxonomy of these bacteria, previously considered distinct species. The most notable synonym is V. alginolyticus E0666, linked to food poisoning and murine virulence. The presence of virulence factors like Ace and Zot, previously noted in V. antiquarius EX25, is conserved across the expanded V. diabolicus subclade, suggesting virulence-associated genes may be ubiquitous within the species and play a fundamental ecological role.
Intrinsic antibiotic resistance in potentially virulent V. diabolicus strains could complicate treatment. Resistome analysis revealed resistance genes for fluoroquinolones, fosfomycin, tetracycline, and penicillin. β-lactamase production and resistance to penicillin, ampicillin, and cephalosporin were experimentally confirmed in V. antiquarius 939. Efflux pumps could further broaden resistance mechanisms. The ubiquity of these resistance features is concerning, particularly given the species’ perceived rarity, mirroring the growing antibiotic resistance trends in V. cholerae, V. parahaemolyticus, and V. vulnificus.
Conclusion
This taxonomic and comparative genomic analysis concludes that V. diabolicus and V. antiquarius should be reclassified as the same species. As V. diabolicus was described earlier, V. antiquarius becomes a taxonomic synonym. V. alginolyticus remains distinct, but misidentified strains E0666, FF273, TS13, and V2 are synonyms of V. diabolicus. The understanding of V. diabolicus species boundaries is evolving, evidenced by the recent public availability of three new V. diabolicus genomes.
Author Contributions
JWT, WN, RP, EA, and MS conceived and designed the research project. JWT, JJT, AM, LP, NE, and DNA conducted the genomic analyses and laboratory experiments. All authors contributed to data interpretation and the writing of the manuscript.
Funding
This study was funded by the National Oceanic and Atmospheric Administration’s (NOAA) Oceans and Human Health Initiative (OHHI), the National Marine Fisheries Service (NMFS), the National Research Council (NRC), the Gordon and Betty Moore Foundation (EVA #537.01), and Texas A&M University-Corpus Christi (TAMU-CC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by numerous colleagues. We thank Chris T. Berthiaume, Rhonda Morales, and Thomas Merrick for sequencing and data analysis assistance, and Drs. Toshio Kodama and Tetsuya Iida and the Pathogenic Microbes Repository at Osaka University for providing V. parahaemolyticus RIMD2210633. We also thank Dr. Daniele Provenzano for manuscript proofreading and Dr. Magesh Thiyagarajan for laboratory supplies.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01893/full#supplementary-material
FIGURE S1 | Phylogenetic tree of the V. harveyi clade.
FIGURE S2 | Phylogenetic tree of β-lactamases.
TABLE S1 | List of the 49 Harveyi clade genomes included in this study.
Footnotes
[1] List of Prokaryotic names with Standing in Nomenclature. http://www.bacterio.net/vibrioc.html
[2] Trim Galore! http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
[3] FigTree version 1.4.3 http://tree.bio.ed.ac.uk/software/figtree/
[4] List of Prokaryotic names with Standing in Nomenclature. http://www.bacterio.net/vibrioc.html
References
[References are in the original article and are linked using markdown syntax]
Keywords: Vibrio diabolicus, Vibrio antiquarius, Vibrio alginolyticus, deep-sea, taxonomy, genomics, diversity, comparative synonym